



| Investigador Principal (IP): Sobrido Gómez, Mª Jesús | Código: PI12/00742 |
| Convocatoria: Proyectos de Investigación en Salud- Instituto de Salud Carlos III (ISCIII)
(año 2012) Ministerio de Ciencia e Innovación |
|
| Presupuesto total: 130.410,00€ | Total concedido: 96.195,00€ |
| Duración: 3 años | Fecha inicio: 01/01/2013 |
| Objeto: El objetivo principal de la investigación propuesta es dar continuidad a una de las líneas de investigación principales de nuestro grupo, las bases moleculares de las ataxias. Gracias a proyectos financiados anteriormente hemos podido identificar que una expansión hexanucleotídica de varias kilobases en el intrón 1 de NOP56 es la causa de un nuevo tipo de ataxia espinocerebelosa (SCA36), en numerosos pacientes gallegos (Garcia-Murias et al., 2012). Pretendemos ahora avanzar en el conocimiento molecular de esta mutación y sus consecuencias. En particular, entender el origen de la mutación, su dinámica en células somáticas y germinales, el efecto sobre la expresión génica y la relación entre el tamaño mutacional y las diversas manifestaciones fenotípicas.Palabras clave: SCA36, ataxia espinocerebelosa, enfermedad de neurona motora, PET, RM, pruebas neurofisiológicas, escalas clínicas, tests neuropsicológicos | |
EQUIPO
| Apellidos | Nombre | Titulación/ Categoría | Entidad |
| Sobrido Gómez | Mª Jesús | Medicina (especialidad Neurogenética), doctor/ IP | Fundación Pública Galega de Medicina Xenómica (FPGMX) |
| Quintáns Castro | Beatriz | Medicina (especialidad Neurología), doctor/ colaborador | Servizo Galego de Saúde (SERGAS) |
| Pérez Gregorio | Alejandra | colaborador | ND |
| Sin especificar | ND | Personal de apoyo (PA) | Detalle en tabla siguiente |
OTRAS ENTIDADES COLABORADORAS
| Nombre | Siglas | Tipo/ Categoría |
| Servizo Galego de Saúde | SERGAS | Organismo público |
| Complejo Hospitalario de Vigo | CHUVI | Hospital público universitario |
| Hospital Fundación Jiménez Díaz, Madrid | FJD | Fundación/ asistencia sanitaria |
| Hospital Clinic, Barcelona | CLINIC | Hospital público universitario |
| Universidad de Hiroshima, Japón(Departamento de Neurociencia Clínica y Terapéutica) | 広島大学 | Universidad pública |
| Universidad de Kyoto, Japón(Departamento de Salud y Ciencias Medioambientales) | 京大 Kyōdai | Universidad pública |
Resumen y justificación del proyecto.
SCA36 es una nueva ataxia espinocerebelosa descrita recientemente por nuestro grupo y otros, causada por un repeat hexanucleotídico en el intrón 1 de NOP56, que se suma a la lista de enfermedades neurodegenerativas causadas por expansiones de DNA y presumiblemente asociadas a desregulación de la transcripción mediada por RNA (véanse figuras 1 y 2).
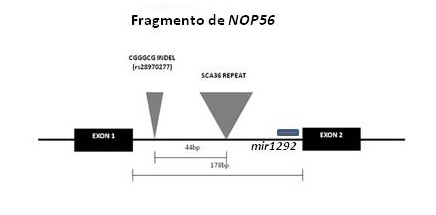
Figura 1
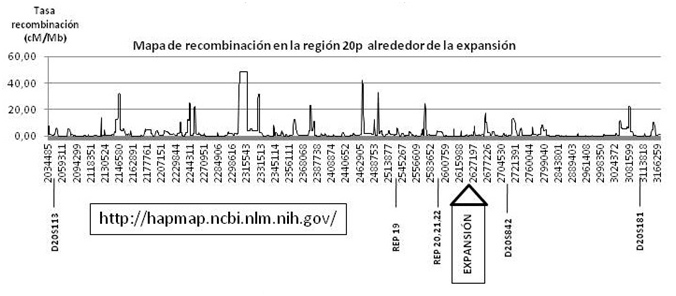
Figura 2
Para comprender los mecanismos moleculares de SCA36 e identificar potenciales estrategias terapéuticas nos planteamos ahora tres bloques de cuestiones:
1) Epidemiología y filogenética de la mutación.
2) Dinámica de la expansión, inestabilidad mitótica y meiótica, influencia en las características fenotípicas.
3) Efectos sobre la transcripción de otros genes.
Para abordar estas cuestiones se planifican las siguientes tareas:
1) Chequeo de SCA36 en otras poblaciones,
2) Reconstrucción filogenética a partir de haplotipos detallados,
3) Comparación del tamaño de la expansión en la transmisión intergeneracional, así como en células somáticas (primarias y transformadas) y germinales (esperma), mediante Southern blot y small-pool PCR,
4) Comparación de las características fenotípicas en función del tamaño mutacional,
5) Estudios de tránscritos candidatos y transcriptoma completo en células primarias (linfocitos, fibroblastos) e inmortalizadas (linfoblastoides).
La investigación genotipo-fenotipo no se limitará a comparar el tamaño de expansión con características generales (edad de inicio, severidad clínica global), sino que se compararán múltiples parámetros clínicos obtenidos en el subproyecto 2. Este nivel de integración será posible gracias a la ontología desarrollada en el subproyecto 3 y servirá para explorar el uso de estas herramientas informáticas en la investigación de la compleja relación genotipo-fenotipo en enfermedades neurodegenerativas.Las interacciones entre subproyectos se representan en la figura 3 a continuación:
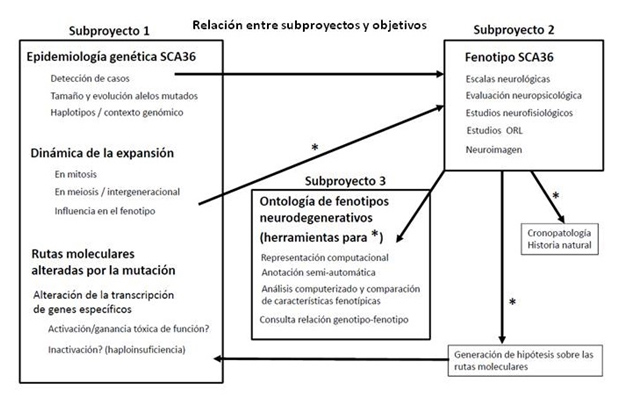
Figura 3
Hipótesis de partida.
Probablemente existieron uno o pocos cromosomas fundadores de SCA36, con un haplotipo de predisposición a la expansión, cuyo origen y filogenia pueden ser estudiados mediante marcadores intragénicos o en la estrecha vecindad. La mutación causante de SCA36 existe en otras regiones de España y del mundo con frecuencias variables debidas a la historia genética poblacional.
La expansión de NOP56 es inestable, tanto en mitosis como en meiosis, lo cual contribuye a explicar la variabilidad clínica en y entre familias. Probablemente el sexo del progenitor transmisor y, tal vez, el sexo del hijo, influyen en esta inestabilidad.
Existe una relación entre el tamaño de la expansión y las características clínicas, que puede ser analizada más adecuadamente si se lleva a cabo una caracterización fenotípica reglada de múltiples parámetros cuantitativos y semi-cuantitativos (subproyecto 2) y si se dispone de las herramientas informáticas para computerizar el análisis fenotípico (subproyecto 3).
La fisiopatología molecular se debe a desregulación mediada por RNA y puede ser estudiada mediante análisis de transcripción en líneas celulares. La expansión intrónica de NOP56 ejerce su efecto deletéreo en la función celular a través de la alteración de expresión de más de un gen, quizá con mecanismos combinados de ganancia tóxica y déficit de función
Entre pacientes gallegos y japoneses hay diferencias fenotípicas que se deben a peculiaridades en las rutas moleculares afectadas. Por ejemplo, pensamos que es probable que el gen TMC2, vecino de NOP56, esté afectado en los pacientes gallegos pero no en los japoneses. Esto podría deberse a la presencia de elementos diferentes en los haplotipos patológicos gallego y japonés, por ejemplo una secuencia aisladora (insulator) entre NOP56 y TMC2 que evitase la inactivación de TMC2 debida a la metilación del repeat hexanucleotídico expandido. La hipoacusia se debería entonces a haploinsuficiencia de TMC2.
Objetivos.
Para este proyecto coordinado, se plantearon inicialmentetres objetivos principales, con sus correspondientes objetivos subordinados:
1 Conocer la epidemiología genética de SCA36:
1.1. Estudiar la frecuencia de SCA36 en pacientes con ataxia espinocerebelosa de otras regiones españolas y otras poblaciones.
1.2. Investigar el origen evolutivo y filogenia de la expansión (mutación fundadora o múltiples eventos).
2 Comprender la dinámica de la expansión intrónica de NOP56 en meiosis, mitosis y su influencia en el fenotipo.
2.1 Estudiar la dinámica de la expansión en mitosis.
2.2. Estudiar la dinámica de la expansión durante la meiosis y transmisión intergeneracional.
2.3. Investigar la relación genotipo-fenotipo.
3 Genómica funcional: investigar el efecto de la expansión SCA36 sobre la transcripción génica.
3.1 Análisis de expresión de genes candidatos
3.2 Estudio del transcriptoma de SCA36 (aproximación sin suposiciones-hypothesis free).
Plan de trabajo planteado y cronograma.
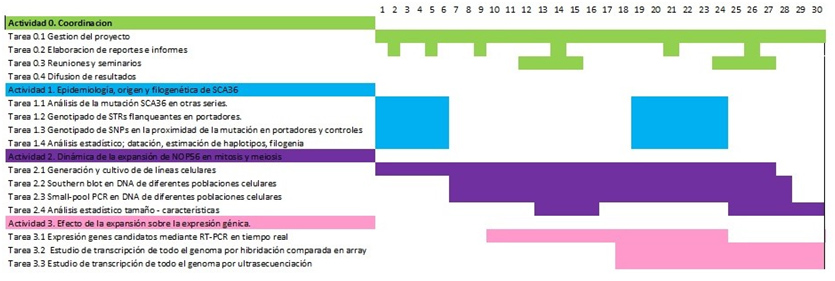
Bibliografía (estado del arte previo a la realización del proyecto).
Armour et al. Direct analysis by small-pool PCR of MS205 minisatellite mutation rates in sperm after mutagenic therapies. Mut Res 1999; 445: 73-80.
Bakalkin et al. Prodynorphin mutations cause the neurodegenerative disorder spinocerebellar ataxia type 23. AM J Hum Genet 2010; 87: 593-603.
De Biase et al. Somatic instability of the expanded GAA triplet-repeat sequence in Friedreich ataxia progresses throughout life. Genomics 2007; 90: 1-5.
DeJesus-Hernandez et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011; 72: 1-12.
Dichgans et al. Spinocerebellar ataxia type 6: evidence for a strong founder effect among German families. Neurology 1999; 52: 849-851.
Excoffier L, Laval G, Schneider S: Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinformatics Online 2005; 1: 47 – 50.
García-Murias et al. Costa da Morte’ ataxia is spinocerebellar ataxia 36: clinical and genetic characterization. Brain 2012, doi:10.1093/brain/aws069
Gaspar et al. Ancestral origins of the Machado-Joseph disease mutation: a worldwide haplotype study. Am J Hum Genet 2001; 68: 523-8.
Gautier et al. Nucleolar KKE/D repeat proteins Nop56p and Nop58p interact with Nop1p and are required for ribosome biogenesis. Molec Cell Biol 1997; 17: 7088-7098.
Gomes-Pereira et al. Analysis of unstable triplet repeats using small-pool polymerase chain reaction. Methods Mol Biol 2004; 277: 61-76.
Felsenstein J. PHYLIP: phylogeny inference package (version 3.2). Cladistics 1989; 5: 164-166.
Kennedy et al. Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum Mol Genet 2003; 12: 3359-3367.
Kobayashi et al. Expansion of Intronic GGCCTG Hexanucleotide Repeat in NOP56 Causes SCA36, a Type of Spinocerebellar Ataxia Accompanied by Motor Neuron Involvement. AJHGenet 201; 89:1-10.
Martorell et al. Progression of somatic CTG repeat length heterogeneity in the blood cells of myotonic dystrophy patients. Hum Mol Genet 1998; 7: 307-312.
Renton et al. A Hexanucleotide Repeat Expansion in C9ORF72 is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011; 72: 1-12.
Sapp et al. Identification of Two Novel Loci for Dominantly Inherited Familial Amyotrophic Lateral Sclerosis. Am J Hum Genet 2003; 73: 397-403.
Todd et al. RNA Mediated Neurodegeneration in Repeat Expansion Disorders.AnnNeur2010;67:291-300.
Verbeek et al. Haplotype study in Dutch SCA3 and SCA6 families: evidence for common founder mutations. Eur J Hum Genet 2004; 12: 441-6.
Wang et al. Prediction of both conserved and nonconserved microRNA targets in animals. Bioinformatics 2008; 24: 325-332.
Wang et al. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genetics 2009; 10: 57-63.
Wang et al. TGM6 identified as a novel causative gene of spinocerebellar ataxias using exome sequencing. Brain 2010; 133: 3510-3518.
Wheeler et al. Length-dependent gametic CAG repeat instability in the Huntington’s disease knock-in mouse. Hum Mol Genet 1999; 8: 115-122.
Zoghbi HY, Orr HT. Pathogenic Mechanisms of a Polyglutamine-mediated Neurodegenerative Disease, Spinocerebellar Ataxia Type 1. J Biol Chem 2009; 284: 7425 -7429.
Estatus actual.
Proyecto en marcha. Fecha finalización prevista 31/12/2015
Publicaciones asociadas (a fecha del 30/09/2013).
WoS: Web of Science <http://www.accesowok.fecyt.es/>
Título: Analysis of the C9orf72 Gene in Patients with Amyotrophic Lateral Sclerosis in Spain and Different Populations Worldwide.
Revista: Human Mutation 2013;34:79-82.
Categoría/Especialidad(WoS):Genetics & Heredity
Factor de impacto (WoS): 5.95
Autores: García-Redondo A, Dols-Icardo O, Rojas-García R, Esteban-Pérez J, Cordero-Vázquez P, Muñoz-Blanco JL, Catalina I, González-Muñoz M, Varona L, Sarasola E, Povedano M, Sevilla T, Guerrero A, Pardo J, de Munain AL, Márquez-Infante C, de Rivera FJ, Pastor P, Jericó I, de Arcaya AA, Mora JS, Clarimón J; The C9ORF72 Spanish Study Group, Gonzalo-Martínez JF, Juárez-Rufián A, Atencia G, Jiménez-Bautista R, Morán Y, Mascías J, Hernández-Barral M, Kapetanovic S, García-Barcina M, Alcalá C, Vela A, Ramírez-Ramos C, Galán L, Pérez-Tur J, Quintáns B, Sobrido MJ, Fernández-Torrón R, Poza JJ, Gorostidi A, Paradas C, Villoslada P, Larrodé P, Capablo JL, Pascual-Calvet J, Goñi M, Morgado Y, Guitart M, Moreno-Laguna S, Rueda A, Martín-Estefanía C, Cemillán C, Blesa R, Lleó A.
Título: SNOMED CT module-driven clinical archetype management
Revista: Journal of Biomedical Informatics Año: 2013 Vol.: 46(3) Págs.: 388-400
Categoría/Especialidad (WoS): Medical Informatics
Factor de Impacto (WoS): 2.131 (posición 06/23)
Categoría/Especialidad (WoS): Computer science, Interdisciplinary applications
Factor de Impacto (WoS): 2.131 (posición 21/99)
Autores: Allones JL, Taboada M, Martínez D, Lozano R, Sobrido MJ
Otras contribuciones (a fecha del 01/08/2014).
Presentaciones en congresos.
Congreso: “XXVII Congreso Nacional de la Asociación Española de Genética Humana”. Madrid (España). Abril 2013.
Título: «¿Deben incluirse las expansiones causantes de ataxia en el algoritmo diagnóstico de las paraparesias espásticas?» (póster).
Autores: Z. Yáñez Torregroza, B. Quintáns Castro, M. García-Murias, A. Ordóñez-Ugalde, I Pascual, L Gómez-Romero, J Arpa, A. Carracedo Álvarez, M. J. Sobrido.
Congreso: “Mutation Detetion 2013”,Lake Louise (Canadá). Abril 2013.
Título: » NGS-elusive mutations: SCA36 illustrates the difficulties to analyze large repeat expansions» (póster).
Autores: García-Murias M, Yáñez-Torregroza Z, Quintáns B, Arias M, Seixas AI, Carneiro I, Silveira I, Carracedo A, Sobrido MJ.
Congreso: “Federación Española de Ataxia (FEDAES)”, Villagarcía de Campos (Valladolid, España). Junio 2013.
Título: “Actualización en ataxias en el congreso SPATAX» (conferencia invitada)
Autores: MJ Sobrido
Congreso: World Federation of Neurology. Viena (Austria), Septiembre 2013.
Título: “Clinical and neuroimaging features of familial C9FTD/ALS: a case report” (póster).
Autores: J Pardo, M Pardo-Parrado, J Clarimón, A Garcia-Redondo, E Cebrián, , I Jiménez-Martínez, J Cortés, P Aguiar, JM Castiñeiras, Tania García-Sobrino, B Quintáns, MJ Sobrido
Congreso: American Society of Human Genetics. Boston (EEUU), Octubre 2013.
Título: “Genetics of spinocerebellar ataxias in Portuguese families: screeningfor SCA15, SCA28 and SCA36” (póster).
Autores: JR Loureiro, AI Seixas, JL Loureiro, A Carracedo, MJ Sobrido, P Coutinho, JSequeiros, I Silveira.
«Spinocerebellar Ataxia Type 36». In: In: Pagon RA, Adam MP, Bird TD, et al., editors. GeneReviews™ [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2013. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1116/ (Manuscrito en preparación).
Autores: Arias M., Quintáns B., García-Murias M., Sobrido MJ.

